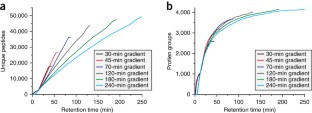
Recent advances in chromatography and mass spectrometry (MS) have made rapid and deep proteomic profiling possible. To maximize the performance of the recently produced Orbitrap hybrid mass spectrometer, we have developed a protocol that combines improved sample preparation (including optimized cellular lysis by extensive bead beating) and chromatographic conditions (specifically, 30-cm capillary columns packed with 1.7-μm bridged ethylene hybrid material) and the manufacture of a column heater (to accommodate flow rates of 350–375 nl/min) that increases the number of proteins identified across a single liquid chromatography–tandem MS (LC-MS/MS) separation, thereby reducing the need for extensive sample fractionation. This strategy allowed the identification of up to 4,002 proteins (at a 1% false discovery rate (FDR)) in yeast (Saccharomyces cerevisiae strain BY4741) over 70 min of LC-MS/MS analysis. Quintuplicate analysis of technical replicates reveals 83% overlap at the protein level, thus demonstrating the reproducibility of this procedure. This protocol, which includes cell lysis, overnight tryptic digestion, sample analysis and database searching, takes ∼ 24 h to complete. Aspects of this protocol, including chromatographic separation and instrument parameters, can be adapted for the optimal analysis of other organisms.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
265,23 € per year
only 22,10 € per issue
Buy this article
- Purchase on SpringerLink
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout

Similar content being viewed by others

Ultra-fast label-free quantification and comprehensive proteome coverage with narrow-window data-independent acquisition
Article Open access 01 February 2024
Robust, reproducible and quantitative analysis of thousands of proteomes by micro-flow LC–MS/MS
Article Open access 09 January 2020

Quantitative shotgun proteome analysis by direct infusion
Article 23 November 2020
References
- Tabb, D.L. et al. Repeatability and reproducibility in proteomic identifications by liquid chromatography-tandem mass spectrometry. J. Proteome Res.9, 761–776 (2010). ArticleCASGoogle Scholar
- Liu, H., Sadygov, R.G. & Yates, J.R. III. A model for random sampling and estimation of relative protein abundance in shotgun proteomics. Anal. Chem.76, 4193–4201 (2004). ArticleCASGoogle Scholar
- Washburn, M.P., Wolters, D. & Yates, J.R. III. Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat. Biotechnol.19, 242–247 (2001). ArticleCASGoogle Scholar
- Hebert, A.S. et al. The one hour yeast proteome. Mol. Cell Proteomics13, 339–347 (2014). ArticleCASGoogle Scholar
- Senko, M.W. et al. Novel parallelized quadrupole/linear ion trap/Orbitrap tribrid mass spectrometer improving proteome coverage and peptide identification rates. Anal. Chem.85, 11710–11714 (2013). ArticleCASGoogle Scholar
- Goffeau, A. et al. Life with 6000 genes. Science274 546 563–547 (1996). ArticleGoogle Scholar
- Ghaemmaghami, S. et al. Global analysis of protein expression in yeast. Nature425, 737–741 (2003). ArticleCASGoogle Scholar
- de Godoy, L.M. et al. Comprehensive mass-spectrometry-based proteome quantification of haploid versus diploid yeast. Nature455, 1251–1254 (2008). ArticleCASGoogle Scholar
- Wu, R. et al. Correct interpretation of comprehensive phosphorylation dynamics requires normalization by protein expression changes. Mol. Cell Proteomics10, M111.009654 (2011). ArticleGoogle Scholar
- Webb, K.J., Xu, T., Park, S.K. & Yates, J.R. III. Modified MuDPIT separation identified 4488 proteins in a system-wide analysis of quiescence in yeast. J. Proteome Res.12, 2177–2184 (2013). ArticleCASGoogle Scholar
- Nagaraj, N. et al. System-wide perturbation analysis with nearly complete coverage of the yeast proteome by single-shot ultra HPLC runs on a bench top Orbitrap. Mol. Cell Proteomics11, M111.013722 (2012). ArticleGoogle Scholar
- Kulak, N.A., Pichler, G., Paron, I., Nagaraj, N. & Mann, M. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nat. Methods11, 319–324 (2014). ArticleCASGoogle Scholar
- Meyer, J.G. & Komives, E.A. Charge state coalescence during electrospray ionization improves peptide identification by tandem mass spectrometry. J. Am. Soc. Mass Spectrom.23, 1390–1399 (2012). ArticleCASGoogle Scholar
- Hahne, H. et al. DMSO enhances electrospray response, boosting sensitivity of proteomic experiments. Nat. Methods10, 989–991 (2013). ArticleCASGoogle Scholar
- Pirmoradian, M. et al. Rapid and deep human proteome analysis by single-dimension shotgun proteomics. Mol. Cell Proteomics12, 3330–3338 (2013). ArticleCASGoogle Scholar
- Huang da, W., Sherman, B.T. & Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc.4, 44–57 (2009). ArticleGoogle Scholar
- Richards, A., Hebert, A., Ulbrich, A., Bailey, D., Coughlin, E., Westphall, M. & Coon, J. Preparation of yeast cells for proteomic analysis by LC-MS/MS. Protocol Exchange (2015) 10.1038/protex.2015.030.
- Treco, D.A. & Winston, F. Growth and manipulation of yeast. Curr. Protoc. Mol. Biol.82, 13.2.1–13.2.12 (2008). ArticleGoogle Scholar
- Elias, J.E. & Gygi, S.P. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods4, 207–214 (2007). ArticleCASGoogle Scholar
- Wenger, C.D., Phanstiel, D.H., Lee, M.V., Bailey, D.J. & Coon, J.J. COMPASS: a suite of pre- and post-search proteomics software tools for OMSSA. Proteomics11, 1064–1074 (2011). ArticleCASGoogle Scholar
Acknowledgements
We are grateful to A. Merrill for yeast production. We thank A. Gasch for assistance with yeast growth. This work was supported by the US National Institutes of Health (R01 GM080148) and the National Science Foundation (0701846). A.L.R. gratefully acknowledges the support from a US National Institutes of Health–funded Genomic Sciences Training Program (5T32HG002760).
Author information
- Alicia L Richards and Alexander S Hebert: These authors contributed equally to this work.
Authors and Affiliations
- The Genome Center of Wisconsin, University of Wisconsin, Madison, Wisconsin, USA Alicia L Richards, Alexander S Hebert, Arne Ulbrich, Derek J Bailey, Emma E Coughlin, Michael S Westphall & Joshua J Coon
- Department of Chemistry, University of Wisconsin, Madison, Wisconsin, USA Alicia L Richards, Arne Ulbrich, Derek J Bailey & Joshua J Coon
- Biomolecular Chemistry, University of Wisconsin, Madison, Wisconsin, USA Alexander S Hebert & Joshua J Coon
- Alicia L Richards
